SARS-CoV-2 (hCoV-19) Genomic Dashboard
enabled by data from 
SARS-CoV-2 genomic sequencing is performed by the WSLH, the Centers for Disease Control and Prevention and a variety of Academic, Public Health, and Contract Laboratories. Due to the time it takes for samples to travel to sequencing laboratories and for the resulting data to be released, it can take on average 2-3 weeks for sequence data to become available after collection from a patient. Additionally, batches of stored samples may often be sequenced at a later date, which can alter historic data. As a result recent trends should be considered preliminary. Only residual positive viral transport media with sufficient viral load from molecular tests can produce reliable sequence data and all of sequence data represented here are from samples collected in Wisconsin.
The SARS-CoV-2 virus like all viruses continues to change over time. As the virus replicates and makes copies of itself, changes in the genome called mutations are introduced. When a virus emerges with one or more new changes it is referred to as a variant of the original virus. These changes in the virus can sometimes lead to a new variant that is better adapted to its environment, which in turn can result in greater spread of the variant. The process of the virus changing over time and the selection of better adapted variants is called virus evolution.
Enabling communication of viral variants requires a process of naming and classifying the variants as they emerge. Several methods for SARS-CoV-2 variant classification have been developed but the most widely adopted is the Pangolin naming scheme, which provides a dynamic naming approach that can adapt to the evolving virus.
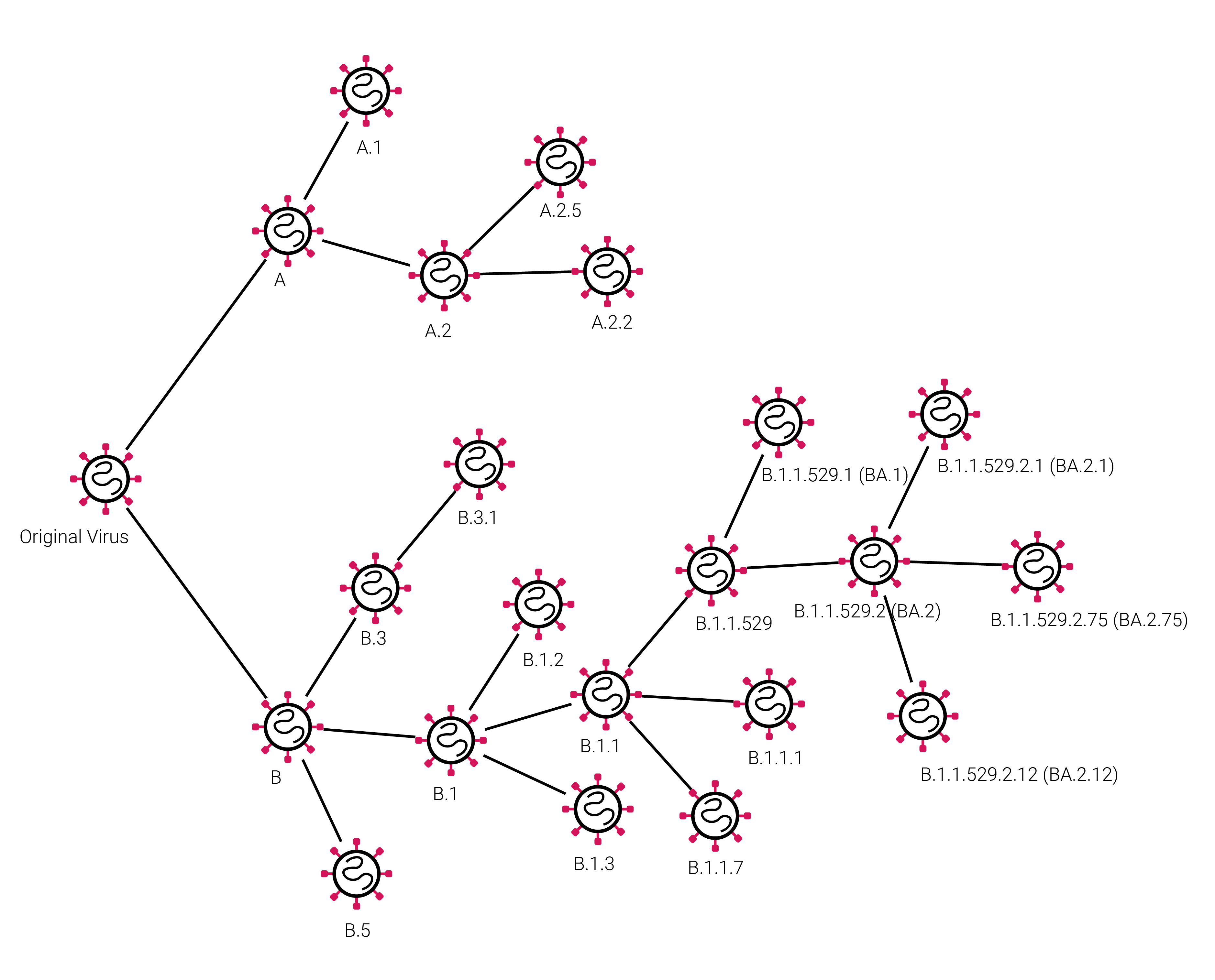
The diagram above depicts the Pangolin naming scheme, starting with the original virus the diagram branches into to branches “A” and “B”. Subsequent viral descendants are given a increasing numerical suffix i.e. “B.1” or “B.1.3”. When the lineage name gets to a length of 5 an alias is given to shorten the overall name. Shown here “BA” is an alias for “B.1.1.529”, which results in viral lineages like “BA.2.75” referencing B.1.1.529.2.75. Note for clarity not all possible lineages are shown in the diagram above.